(1)
Department of Mathematics, The Univeristy of Hong Kong
(2) Department of Computer Science, The University of Hong Kong
(3) Genome Research Center, The University of Hong Kong
Cluster and set-cover algorithms are developed to obtain a set of tag SNPs that can represent all the known SNPs in a chromosomal region, subject to the constraint that all SNPs must have a squared correlation R2 > C with at least one tag SNP, where C is specified by the user. The program is implemented with Java, and it can run in Windows platform as well as the Unix environment.
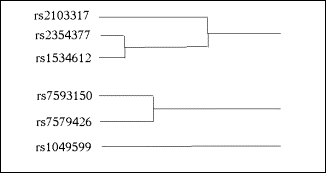
Hierarchical clustering starts with a square matrix of pair-wise distances between the objects to be clustered. For the problem of tag SNP selection, the objects to be clustered are the SNPs, and an appropriate measure of distance is 1-R2, where R2 is the squared correlation between two SNPs. The rationale is this: the required sample size for a tag SNP to detect an indirect association with a disease is inversely proportional to the R2 between the tag SNP and the causal SNP. In agglomerative clustering, the two clusters with the smallest inter-cluster distance are successively merged until all the objects have been merged into a single cluster. Different forms of agglomerative clustering differ in the definition of the distance between two clusters, each of which may contain more than one object. In single-linkage or nearest-neighbour clustering, the distance between two clusters is the distance between the nearest pair of objects, one from each cluster. In complete linkage or farthest neighbour clustering, the distance between two clusters is the distance between the farthest pair of objects, one from each cluster. The clustering process can be represented by a dendrogram, which is a tree diagram showing how the individual objects are successively merged at greater distances into larger and fewer clusters. The user defines a cutoff merging distance, and considers all the distinct clusters that have been generated at that distance or lower (see Figure 1).
A desirable property for a clustering algorithm, in the context of tag SNP selection, would be that a cluster must contain at least one SNP (the tag SNP) that is no more than the merging distance from all the other SNPs from the same cluster. If this is the case, then by setting a cutoff merging distance of C, one can ensure that no SNP is further than C away from the tag SNP in its cluster. In this sense, neither of the methods proposed by Byng et al (2003) is ideal, since the single-linkage method does not guarantee the existence of a tag SNP with distance less than C from all SNPs in the same cluster, while complete-linkage is too conservative in that all SNPs have distance under C from all other SNPs in the same cluster.
In order to
achieve the desired property described above, we propose a new definition of the
distance between two clusters that, as follows:
*
For
each SNP belonging to either cluster, find the maximum distance between it and
all the other SNPs in the two clusters
*
The
smallest of these maximum distances is defined as the distance between the two
clusters.
*
The corresponding SNP is defined as the
tag SNP of the newly merged cluster
We call this method minimax clustering. Similar algorithms have been described for the selection of compounds with diverse properties (Snarey et al, 1997). There is a parrallel in topology in which the distance between two compact sets can be measured by a sup-inf metric known as Hausdorff distance (Barnsley, 1988).
We have compared the performance of the three implemented algorithms, using SNP data from the ENCODE regions of the HapMap project, according to three criteria: (1) compression, the ratio of clusters to SNPs, (2) compactness, the average distance between a SNP and the tag SNP of its cluster, and (3) run time. Our results show that the compression ratio is roughly equivalent for the set cover and minimax clustering algorithms but substantially higher for the complete linkage (Table 1). The minimax algorithm produces more compact clusters than the set cover algorithm, but takes approximately twice the running time. Further evaluations of these and other algorithms for tag SNP selection are needed.
Fig. 1. Sample illustrative dendrogram showing how 7 SNPs are merged into 3 clusters at or below the cutoff merging distance.
Table 1. Properties of three tag SNP selection
algorithms, evaluated for ENCODE regions.
|
Encode Region |
Compression |
Compactness |
Run Time
(seconds) | ||||||
|
Complete |
Minimax |
Set cover |
Complete |
Minimax |
Set cover |
Complete |
Minimax |
Set cover | |
|
2A |
0.276 |
0.243 |
0.245 |
0.021 |
0.033 |
0.037 |
3.94 |
5.42 |
3.20 |
|
2B |
0.289 |
0.254 |
0.259 |
0.018 |
0.033 |
0.032 |
5.44 |
6.92 |
4.03 |
|
4 |
0.241 |
0.209 |
0.208 |
0.016 |
0.031 |
0.035 |
6.53 |
13.30 |
5.25 |
|
7A |
0.312 |
0.278 |
0.278 |
0.013 |
0.028 |
0.032 |
2.56 |
3.39 |
2.00 |
|
7B |
0.184 |
0.164 |
0.168 |
0.020 |
0.030 |
0.035 |
3.53 |
5.03 |
2.84 |
|
7C |
0.238 |
0.215 |
0.212 |
0.018 |
0.019 |
0.021 |
2.38 |
3.28 |
1.80 |
|
8A |
0.266 |
0.242 |
0.242 |
0.019 |
0.035 |
0.040 |
2.39 |
2.94 |
1.83 |
|
9 |
0.357 |
0.314 |
0.310 |
0.012 |
0.025 |
0.031 |
1.47 |
1.74 |
0.98 |
|
12 |
0.258 |
0.225 |
0.225 |
0.017 |
0.028 |
0.034 |
2.69 |
3.69 |
2.03 |
|
18 |
0.280 |
0.251 |
0.251 |
0.014 |
0.033 |
0.037 |
2.17 |
2.81 |
1.64 |
Table 2. Comparison of three tag SNP selection
algorithms, evaluated for ENCODE
chromosome 9
region
for different threshold values.
|
Threshold
Values |
Compression |
Compactness |
||||
|
Complete |
Minimax |
Set cover |
Complete |
Minimax |
Set cover |
|
| 0.70 | 0.314 | 0.291 | 0.287 | 0.026 | 0.042 | 0.043 |
|
0.75 |
0.333 | 0.302 | 0.298 | 0.020 | 0.036 | 0.035 |
|
0.80 |
0.360 |
0.318 |
0.314 |
0.012 |
0.025 |
0.031 |
|
0.85 |
0.384 | 0.349 | 0.349 | 0.010 | 0.0145 | 0.0151 |
|
0.90 |
0.430 | 0.368 | 0.368 | 0.0057 | 0.0117 | 0.0117 |
|
0.95 |
0.492 | 0.473 | 0.473 | 0.0014 | 0.0018 | 0.0017 |
The program is available for free download.
Option 1: Click here if one would like to download the original version TaggingSetChooser.jar and mail.jar.
Option 2: Click here for downloading version 2 TaggingSetChooser_v2.jar and mail.jar. Put them into the same directory and save the TaggingSetChooser_v2.jar file as TaggingSetChooser.jar. Version 2 is an implementation that can handle the case when the input correlation matrix is a band matrix. Band matrix can be produced by programs like Haploview etc., when the option of computing correlation between SNPs within a certain kB region is set. With the band matrix, less correlation pairs are needed during the clustering process and we have tested with the ENCODE data that a faster speed can be achieved. It should be noted that, even though in our test data, these two versions produce very similar clusters, in the band version, there are sometimes the needs to have one or two more tag SNPs in order to tag all SNPs. The version 2 of band matrix capability can also work well with the full correlation matrix and we have tested that, on that case, the outputs of these two versions are the same (read Tagging results of chromosome 8, 9, 18 of full r vs band r.doc).
Sample program outputs for the minimax clustering are available here: the html output, the html members output and the text format output.
The sample DOS script is available for download. An user has helpfully suggested the script for Mac and Linux for download.
The path "data" and "results" can be changed. In case of no change, then the user should create these two folders and the "temp" folder under the directory containing the program file. The file names *.txt, *.members and *.out can also be set by the users. The parameters "threshold" and the "scale" can be set by the users. The "threshold" is for the threshold value used by the algorithms and the "scale" is for the scale of the html output.
The program requires two input files, one for the LD relationship between each SNPs pair and the other for each SNP's position and MAF data. The sample LD file can be download here and the sample MAF file here (in ascending order of the SNP position). The data columns in the input files are separated by one space " ". And the LD data can be generated, for example, with the Haploview.
And, here is the zipped file of the CLUSTAG v2 with all the sample flies, the necessary directories and the DOS script, so that the user can run the program after unzipping this file. The team of P. Sham have developed a program that have the similar role of the Haploview, and a script (run_programs.bat) has been written to run both this pre-processing program and the CLUSTAG. These programs (along with Clustag v2), readme file and sample input files can be download here as a zipped file.
Here are the questions and answers page for some users who have encountered some common questions with CLUSTAG.
The ENCODE data were downloaded from HAPMAP's site www.hapmap.org on Jun 30, 2004 and is based on
NCBI build3. The work is supported by a small project grant from the University of Hong Kong.Barnsley M. F. (1988) Fractals everywhere. Academic Press.
Barrett,
J.C., et al. (2004) Haploview: Analysis and Visualization of LD and Haplotype
Maps. Bioinformatics, Advance Access.
Byng,
M., et al. (2003) SNP Subset Selection for Genetic Association Studies. Annals.
Of Human Genetics, 67, 543-556.
Carlson,
C., et al. (2004) Selecting a Maximally Informative Set of Single-Nucleotide
Polymorphisms for Association Analyses Using Linkage Disequilibrium. Am. J. Hum.
Genet. 74:106-120.
Johnson, G., et
al. (2001) Haplotype Tagging
for the Identification of
Common
Disease
Genes.
Nat Genet 29(2):233-7
Reuven,
Y., and Zehavit, K. (2004) Approximating the Dense Set-Cover Problem. J.
Computer and System Sciences. In Press.
Snarey,
M., Terrett N., Willett P. and Wilton D. (1997) Comparison of Algorithms for
Dissimilarity-Based Compound Selection. J. Molecular Graphics and Modelling,
15(6), 372-385.
Note on Clustering Methods: Two more papers of the clustering methods and their comparison for the gene expression data are listed below:
Mcshane L.M., Radmacher M.D., Freidlin B., Yu R., Li M.C., and Simon R. (2002) Methods for assessing reproducibility of clustering patterns observed in analyses of microarray data. Bioinformatics. v11, 1462-9.
Datta S. and Data S. (2003) Comparisons and validation of statistical clustering techniques for microarray gene expression data. Bioinformatics. v19, 459-466.
Note on the minimax algorithm: It is pointed out that there exists a parallel from topology with the minimax algorithm. It is the Hausdorff distance, which measures the maximum distance of a set to the nearest point in the other set. More details can be found at:
Rucklidge W. (1996) Efficient visual recognition using the Hausdorff distance. Springer.
Note on Run Time: The complexity of the clustering methods are of order O(n2). With the run time information in our table of several hundred SNPs and this complexity information, the users can estimate roughly the expected run time for their samples before the program's execution. The run time will not be an issue for data of several hundred to a hundred thousand SNPs. But, it will be a constraint when we are studying the whole genome at one time, when the size may be of several million SNPs. This is an area of further work as the HAPMAP project is producing the whole genome haplotype information.
Note on Graph-Method Algorithm: Two more papers from different journals are cited, which is about the graph-method algorithm. The one by Johnson (1973) studied the error bound of the algorithm and the one by Fujito (2001) studied the case of weighted edges.
Johnson D. (1973) Approximation Algorithms for
Combinatorial Problems. Annual ACM Symposium on Theory of Computing. 38-49.
Fujito T. (2001) On approximability of the independent/connected edge dominating
set problems. Information Processing Letters. v79, 261-266.
Note on the CLUSTAG: The result is published on:
S. I. Ao, Kevin Yip, Michael Ng, David Cheung, Pui-Yee Fong, Ian Melhado, Pak C. Sham (2005) CLUSTAG: Hierarchical Clustering and Graph Methods for Selecting Tag SNPs. Bioinformatics. v21(8), 1735-1736.
*To whom correspondence should be addressed.